Michelly Yanay Rocobado-Pozo ¹ , María Ruiz-Medina ¹ , Ismael Reina-Reina1, Araceli Reyes-Molina ¹ , José Javier Durán-Ávila ¹
1 Estudiante del Grado en Medicina de la Universidad de Granada (UGR)
TRANSLATED BY:
Celia Manzano-Cruz ² , Lorena Trujillos-Yévenes ² , Alba Casillas-Sánchez ² , Aimar Martínez-López ² , Isabel Barberá- Rodríguez ² , José Manuel Fernández-Fuentes ²
2 Student of the BA in Translation and Interpreting at the University of Granada (UGR)
There are numerous therapeutic alternatives for the treatment of Parkinson’s disease (PD). However, none of this can stop the progression of this disease, although they do have major symptomatic benefit. Thus, interest arises in new therapeutic alternatives such as cell transplantation, glial cell line-derived neurotrophic factor (GDNF) infusion, cell therapy or gene therapy. This review presents a general overview of current therapies used for PD and future lines of research, highlighting the role of gene therapy. The different gene therapy approaches for PD are revised. These include a symptomatic gene therapy, which aims to restore dopamine levels in the brain, and a therapy focused on the modification of the disease, for which the glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) plays a fundamental role. Finally, the authors conclude that these lines of research are still far from clinical application.
Keywords: glial cell line-derived neurotrophic factor, cell therapy, gene therapy, glial cells.
- Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease. It is a pathology in which the nigrostriatal pathway is affected, with a characteristic deficit of dopamine production. PD is characterized by motor symptoms such as freezing, postural instability, rigidity, or tremor, as well as non-motor symptoms, including anxiety, dementia and depression. In this review, the existing therapies for PD are described, with special emphasis on gene therapy, which has promising results to date. In gene therapy, viral vectors are employed to deliver genetic material that will modify the cause of the disease.
- Therapeutic action for Parkinson’s disease
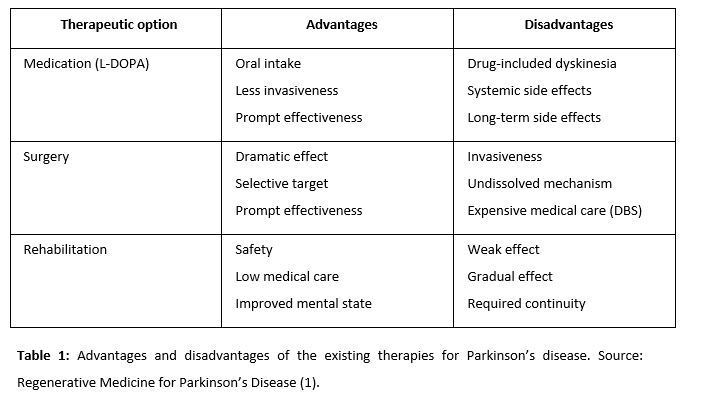
The basis of the pathogenesis of PD is progressive degeneration of dopaminergic neurons in the nigrostriatal system. This makes PD a good target for regenerative medicine (1).
2.1. Historical Development of Regenerative Medicine for PD
2.1.1. Fetal nigral cell transplantation
Fetal nigral cells were transplanted to make new synaptic connections and to synthesize dopamine (2). At the outset of the 21st century, studies revealed insufficient functional recovery and increased dopamine uptake in elderly patients and some patients with tardive dyskinesia. Other studies reported more optimistic aspects of fetal nigral cell transplantation using a cell suspension rather than solid tissue. Patients showed improvement in motor function and a reduced immune response in dyskinesia. Thus, fetal nigral cell transplantation might exert strong therapeutic effects for a long time with appropriate patient selection.
2.1.2. Infusion of glial cell line-derived neurotrophic factor (GDNF)
Alternatively, the intraparenchymal injection of GDNF can have important therapeutic effects, especially if the injection is at the intrastriatal level (3). was demonstrated as another hopeful therapeutic option for PD patients. Moreover, unilateral GDNF administration has been proved to improve bilateral function.
2.1.3. Cell therapy
One cell-based strategy is to transplant encapsulated cells, which allows combining several genetically modified cell types. Cells inside the capsule survived for up to 6 months in vivo with enough oxygen, nutrients, and a semi-permeable membrane. The capsule protects cells from immunological rejection and tumor formation. Neural stem cells (NSCs) and mesenchymal stem cells (MSCs) proved to be good candidates for this therapy, as the number of NSCs that survived in vivo was higher if they had previously been treated with GDNF. GDNF treatment increased the proportion of dopaminergic neurons in MSCs. NSCs were mediated by secreted trophic factor. Embryonic stem cells (ESCs), the last alternative, induce neurons with a high proportion of dopaminergic neurons (4).
2.1.4. Gene therapy
Gene therapy is classified into two groups: in vivo and ex vivo. Direct gene delivery using vectors is the main characteristic of in vivo gene therapy. Herpes simplex virus (HSV), retrovirus, adenovirus, and other non-viral vectors such as liposome are used (5).
The purposes of gene therapy are to increase local dopamine concentration, to exert neuroprotective/neurorestorative effects, to ameliorate the microenvironment of the dopaminergic system involved in PD, and to normalize genetically abnormal cells.
- Biological basis of gene therapy
Around 80% of patients with PD are considered as idiopathic, whereas the remaining 20% cases are presumed to be genetic. In this section, the different genetic factors that influence the pathogenesis of PD are presented.
PD is a neurodegenerative disorder and the most common synucleinopathy (7). It is characterized by the accumulation of anomalous alpha-synuclein aggregates in the cytoplasm of neurons and glial cells. Alpha-synuclein is a fundamental component of Lewy bodies, which play a significant role in the progression of PD. It is believed that alpha-synuclein oligomers and fibrils are toxic. The protein has “prion-like” properties that can spread throughout a network of neurons. This explains the progression of the disease from the basal ganglia to the neocortex. Moreover, alpha-synuclein aggregates with beta-amyloid and tau. However, mutations responsible for the disease were identified in the alpha-synuclein gene. Highly penetrant mutations producing rare, monogenic forms of the disease have been discovered in singular genes such as SNCA. The first identified missense mutation, p.A53T is the most frequent (9), the rest are p.A30P, pE46K, p.H50Q, p.G50D and p.G51D. In addition to SNCA, autosomal-dominant PD-causing mutations with pleomorphic pathology and incomplete penetrance have been found in the gene encoding Leucine-rich repeat kinase 2 (LRRK2) (10). Six mutations in LRRK2 have been identified (five types of missense mutation and one splicing mutation) (11). This protein is involved in numerous cellular processes including autophagy, cytoskeletal dynamics, kinase cascades, mitochondrial function and vesicular trafficking. Overall, mutations in LRRK2 are the most commonly known genetic cause of late-onset familial PD. Although the role of LRRK2 in cellular physiology is still unclear, this recent finding allows for a better understanding of the pathogenesis of PD.
Recently, mutations were also identified in the VPS35 gene, responsible for autosomal dominant late-onset PD. The mutant VPS35 protein alters the trafficking of cathepsin D, a protein involved in the degradation of alpha-synuclein. VPS35 is linked to the other autosomal dominant PD genes SNCA and LRRK2 through endosomes and vesicular trafficking. Mutations in genes PARK2, PINK1, DJ-1, ATP13A2, PLA2G6 and FBX07 have been shown to cause autosomal recessive PD and early-onset. Mutations in PARK2 are the most frequent I of autosomal recessive and early-onset PD due to a microdeletion of D6S305 (12). These mutations consist of exon rearrangements, including both deletions and duplications. The pathology caused by PARK2 consists of severe neuronal loss in the substantia nigra.
Mutations in PINK1 are the second most common cause of autosomal recessive PD (13). The PARK6 locus was first mapped on chromosome 1p36 and mutations in PINK1 are associated with this chromosome. PINK1 is located in the mitochondria of cells and it helps protect mitochondria from malfunctioning during periods of cell stress. When this function is altered, cellular stress occurs, which seems to be related to the development of familial PD. PINK1 phosphorylates PARKIN2 to regulate mitophagy of damaged mitochondria.
Mutations producing rare forms of the disease have been discovered in gene DJ-1. This gene has been reported to protect dopaminergic cells against oxidative stress and to play a key role in maintaining normal dopaminergic function in the nigrostriatal pathway (14). Furthermore, DJ-1 seems to have chaperone activity and the ability to inhibit alpha-synuclein aggregation. It may be involved in transcriptional regulation of neuroprotective and anti-apoptotic genes and in the protection of neurons from oxidative stress induced apoptosis.
Other rare, recessively inherited forms of PD are caused by mutations in three genes: ATP13A2, PLA2G6, y FBXO7. Mutations in GBA1, the gene encoding for the lysosomal enzyme glucocerebrosidase, cause Gaucher disease. Patients suffering from this disease have higher risk of developing PD and have symptoms associated with idiopathic PD. It was observed that mutation in gene GBA1 lead to a loss of beta-glucocerebrosidase activity, which causes lysosomal dysfunction and an increased substrate glucosylceramide accumulation. This led to review the link between the two diseases and the factors that cause monomeric alpha-synuclein to transform into neurotoxic forms and oligomeric aggregates that contribute to the progression of the disease (15).
Finally, it is worth noting the importance of the glial cell line-derived neurotrophic factor (GDNF) and neurturin for the survival of neuronal cells. These, as well as their receptors, were decreased and deregulated in patients with PD (9).
- Gene therapy
Gene therapy is a therapeutic technique in which a functional gene is inserted into a patient’s cells. Its aim is to correct a genetic defect that often causes pathology. It can be performed by using viruses that carry these therapeutic molecules (16). Nowadays, there are two different gene therapy methods: symptomatic gene therapy and gene therapy focused on disease modification (17). Symptomatic gene therapy mainly refers to the transport of the dopa-decarboxylase enzyme, AADC (which transforms levodopa into dopamine). This increases dopamine in the brain, which is deficient in patients with PD (18). This has been tested in animals where lower doses of levodopa meant higher doses with gene transport of AADC, producing an antiparkinsonian effect. Lentiviruses have also been used to transport tyrosine hydroxylase, AADC, and GTP cyclohydrolase, the combination of which results in dopamine. The combination of lentiviruses and dopamine has been tested in animals also having an antiparkinsonian effect.
The second method focuses on GDNF and neurturin. The gene transport of these trophic factors, such as neurturin, protects the nigrostriatal pathway and the motor function. Nonetheless, this was not tested in humans but only in rodents and primates. Quite significantly, the gene transport was highly efficient in preclinical tests, but it was very inefficient when applied to PD patients. This could be due to the extensive degeneration of the nigrostriatal system during treatment. Furthermore, the remaining nerve fibers are defective in the axonal transport mechanisms, so trophic factors are not carried efficiently (19).
Gene therapy in PD is still a mystery, as no study has yet shown undisputed clinical benefit in humans. Although a solution is possible, currently gene therapy has only been used to treat motor symptoms. However, other symptoms such as psychiatric ones would require a different type of gene therapy.
- New therapeutic alternatives
Regarding the treatment of PD, the latest advances propose an imitation of nature. The distribution of neurotrophic factors using retroviruses is a very positive alternative to provide our target cells with these factors.
In this review we intend to address the main therapies for the treatment of PD. It should be noted that, unlike the treatments introduced so far, this new perspective aims to act on the pathogenesis of the disease in order to find an effective treatment instead of a merely symptomatic one.
Neurotrophic factors are biological compounds that stimulate cell regeneration and, therefore, ease the healing process. They function by binding to cellular receptors and subsequently activating the cell signaling cascades that regulate mitosis, differentiation, and apoptosis (20).
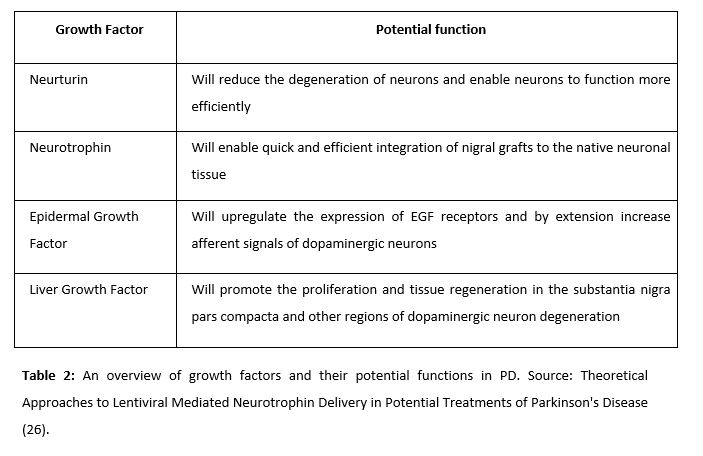
Neurturin (NRTN) is a natural analogue of neurotrophic factors derived from the glial cell line (GDNFs), commonly used to attack dopaminergic neurons in PD animals (21). Its observed effects include delayed degeneration of neurons, selective protection of dopaminergic neurons, and general improvement of neural functioning. The administration of this factor was tested by Gasmi et al in 2007. Animal trials were conducted to achieve striatal delivery of NRTN by using CeRe-120 (an adeno-associated virus type 2 – AAv2). Since then, the administration of this factor has been a very promising alternative for the treatment of PD.
NRTN has been proved to have beneficial effects in idiopathic PD, thanks to its integrating effect of grafts in the substantia nigra (22).
Iwakura’s studies revealed that there was a deficit in the expression of EGF in post-mortem brains of patients with PD. Thus, it was thought that the administration of this factor would be a promising alternative in the treatment of the disease (23). Gobernado et al. administered LGF in rats and observer a possible neuroprotective activity. LGF is a hepatic mitogen that promotes proliferation of various cell types and facilitates tissue regeneration. Upon peripheral application of LGF to the left striatum, sprouting of tyrosine hydroxylase-positive terminals and dopamine transporter expression was unilaterally increased. LGF also stimulated the phosphorylation and regulation of proteins critical for cell survival – including Bcl2 and Akt.
The aim of the administration of genes based on retroviruses is to deliver genes to the target cells, using viruses as means of transport. In this case, lentiviruses are used (25). Once the target genes are packaged into these viral vectors, they convert their single stranded RNA into a double stranded DNA that can stably integrate into the host genome. The integrated vector, called provirus, undergoes replication and transcription in the host genome, producing both viral mRNAs and packaged RNA. The integrated vector, called the provirus, undergoes replication and transcription in the host genome producing the viral mRNAs and the packaged RNA as well.
The importance of controlling the not-so-noble use of these systems, such as overexpression of genes, should be emphasized.
- Conclusion
Today, we can classify PD as a pathology with a multifactorial etiology and a strong genetic influence. Nowadays, the existing therapies focus almost exclusively on the symptomatology. However, recent studies on gene therapy seek to provide an alternative that goes directly to the root of the problem. Through the arguments exposed in this review, it can be concluded that gene therapy may be beneficial for the treatment of PD, although not all the articles reviewed conclude this. There are still numerous aspects to be explored in the field of genetics, which appears to indicate a promising future for the treatment of PD.
Conflicts of Interest Statement
The authors declare that there are no conflicts of interest in this article.
References
- Yashura T, Kameda M, Agari T, Date I. Regenerative Medicine for Parkinson’s Disease. Neurol Med Chir. 2015; 55: 113-123.
- Lindvall O, Rehncrona S, Gustavii B, et al. Fetal dopamine-rich esencephalic grafts in Parkinson’s disease. Lancet. 1998; 2: 1483–1484.
- Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med. 2005; 11: 703–704.
- Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000; 28: 31–40.
- Yasuhara T, Date I. Gene therapy for Parkinson’s disease. J Neural Transm Suppl. 2009; 73: 01–309.
- Bhat S, Acharya UR, Hagiwara Y, Dadmehr N, Adeli H. Parkinson’s disease: Cause factors, measurable indicators, and early diagnosis. Comput Biol Med. 2018; 102: 234–41.
- Toulorge D, Schapira AHV, Hajj R. Molecular changes in the postmortem parkinsonian brain. J Neurochem. 2016; 139: 27–58.
- Schulz JB, Hausmann L, Hardy J. 199 years of Parkinson disease – what have we learned and what is the path to the future? J Neurochem. 2016; 139: 3–7.
- Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: Mendelian versus non‐Mendelian inheritance. J Neurochem. 2016; 139: 59–74.
- Weisenhorn DMV, Giesert F, Wurst W. Diversity matters – heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s Disease. J Neurochem. 2016; 139: 8–26.
- Goedert M, Jakes R, Anthony Crowther R, Grazia Spillantini M. Parkinson’s Disease, Dementia with Lewy Bodies, and Multiple System Atrophy as alpha-Synucleinopathies. Methods Mol Med. 2001; 62: 33–59.
- Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004; 44: 601–607.
- Matsumine H, Yamamura Y, Hattori N, et al. A microdeletion of D6S305 in a family of autosomal recessive juvenile parkinsonism (PARK2) Genomics. 1998; 49: 143–146.
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004; 304: 1158–1160.
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003; 299: 256–259.
- Man JHK, Groenink L, Caiazzo M. Cell reprogramming approaches in gene- and cell-based therapies for Parkinson’s disease. J Control Release. 2018; 286: 114–24.
- Kordower JH, Bjorklund A. Trophic factor gene therapy for Parkinson’s disease. Mov Disord. 2013; 28(1): 96-109.
- Mittermeyer G, Christine CW, Rosenbluth KH, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther. 2012; 23(4): 377-381.
- Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136(Pt 8). 2013; 136(Pt 8): 2419–2431.
- Barker RA. Parkinson’s disease and growth factors — are they the answer?. Parkinsonism Relat Disord. 2009; 15(Suppl 3): 181–184.
- Ye M, Wang XJ, Zhang YH, et al. Transplantation of bone marrow stromal cells containing the neurturin gene in rat model of Parkinson’s disease. Brain Res. 2007; 1142: 206–216.
- Mogi M, Togari A, Kondo T, et al. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1999; 270(1): 45–48.
- Iwakura Y, Piao YS, Mizuno M, et al. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: Neurotrophic implication in nigrostriatal neurons. J Neurochem. 2005; 93(4): 974–983.
- Gonzalo-Gobernado R, Calatrava-Ferreras L, Reimers D, et al. Neuroprotective Activity of Peripherally Administered Liver Growth Factor in a Rat Model of Parkinson’s Disease. PLoS One. 2013; 8(7): 67771.
- Palfi S, Gurruchaga JM, Lepetit H, et al. Long-Term Follow-Up of a Phase I/II Study of ProSavin, a Lentiviral Vector Gene Therapy for Parkinson’s Disease. Hum Gene Ther Clin Dev. 2018; 29(3): 148–55.
- Qudrat A, Unni N. Theoretical Approaches to Lentiviral Mediated Neurotrophin Delivery in Potential Treatments of Parkinson’s Disease. Yale J Biol Med. 2016; 89(2): 215–25.