Michelly Yanay Rocobado-Pozo ¹ , María Ruiz-Medina ¹ , Ismael Reina-Reina1, Araceli Reyes-Molina ¹ , José Javier Durán-Ávila ¹
1 Estudiante del Grado en Medicina de la Universidad de Granada (UGR)
TRANSLATED BY:
Celia Manzano-Cruz ² , Lorena Trujillos-Yévenes ² , Alba Casillas-Sánchez ² , Aimar Martínez-López ² , Isabel Barberá- Rodríguez ² , José Manuel Fernández-Fuentes ²
2 Student of the BA in Translation and Interpreting at the University of Granad
Resumen
La enfermedad de Parkinson (EP) cuenta en la actualidad con un gran número de alternativas terapéuticas, pero ninguna de ellas es capaz de llegar a la curación por sí misma, aunque sí tienen una importante repercusión sintomática. Así surge el interés por nuevas alternativas terapéuticas como el trasplante celular, la infusión del Factor Neurotrópico Derivado de la Glía (GDNF), la terapia celular o la terapia génica. En esta revisión se pretende dar una mirada general tanto de las terapias que actualmente existen como de las presentes y futuras líneas de investigación, destacando el papel de la terapia génica. Posteriormente, se revisan las diferentes variantes que podemos encontrar dentro de la terapia génica aplicada a la EP, tanto una terapia génica sintomática, aumentando la cantidad de dopamina en el cerebro, como una terapia génica centrada en la modificación de la enfermedad, para la que es fundamental la familia de ligandos de las células gliales. Debemos dejar claro, también, que estas líneas de investigación están aún muy lejos de aplicación clínica.
Palabras clave: factor neurotrópico derivado de la glía, terapia celular, terapia génica, células gliales
Keywords: glial cell line-derived neurotrophic factor, cell therapy, gene therapy, glial cells.
1. Introducción
La enfermedad de Parkinson (EP) es la
segunda enfermedad neurodegenerativa más común después de la enfermedad de
Alzheimer. Es una patología en la que se ve afectada la vía nigroestriatal, con
un característico déficit de producción de dopamina. En esta enfermedad
encontramos síntomas motores tales como la congelación, la inestabilidad
postural, la rigidez o el temblor y síntomas no motores como la ansiedad, la
demencia o la depresión. Se van a abordar las distintas terapias existentes en
la actualidad, haciendo especial hincapié en la terapia génica, que tiene
resultados esperanzadores hasta la fecha. Dentro de esta tienen un importante
papel los vectores virales que son capaces de transmitir genes que modificarán
el curso de la enfermedad.
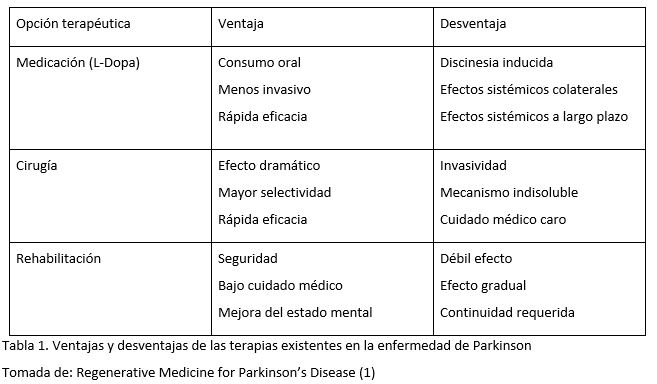
2. Alternativas terapéuticas de la enfermedad del Parkinson
Como bien es sabido, gran parte de la
patología de la EP es la degeneración de las neuronas dopaminérgicas en el sistema nigroestriatal, por lo
que la podemos considerar como una buena diana para la medicina regenerativa
(1).
Desarrollo histórico de la
regeneración médica para la EP:
I. Trasplante de células fetales de la sustancia negra
El trasplante de células fetales de la sustancia negra se realizó con el objetivo de conseguir formar nuevas
conexiones sinápticas, así como la síntesis de dopamina (2). A comienzos de siglo XXI
estudios revelaron una recuperación insuficiente en pacientes ancianos y en
algunos pacientes con discinesia tardía, pero un mayor consumo de dopamina.
Estudios posteriores, realizados esta
vez en suspensión celular en lugar de un tejido sólido, mostraron una mejora en
la función motora, en la discinesia, una menor reacción inmune. De esta manera
se concluyó que este trasplante puede tener fuertes efectos terapéuticos en pacientes bien seleccionados.
II. Infusión del Factor Neurotrópico Derivado de la Glía (GDNF)
Un tratamiento alternativo para
pacientes con EP es la inyección intraparenquimatosa de GDNF, que puede tener importantes efectos terapéuticos, especialmente si la inyección es a
nivel intrastriatal (3). Si la inyección de GDNF es unilateral ha demostrado
mejorar la función bilateral.
III. Terapia celular
Una estrategia celular es el
trasplante de células
encapsuladas, que permite combinar varios tipos celulares, genéticamente modificados.
Estas células han sido capaces de sobrevivir hasta 6
meses in vivo con suficiente oxígeno,
nutrientes y una membrana semipermeable. Esta cápsula previene rechazos
inmunológicos y formaciones tumorales. Tanto células madre mesenquimales (MSCs) como células madre neuronales (NSCs) parecen ser
buenas candidatas para la terapia, demostrándose que el número de células que sobrevivían in vivo era mayor si previamente se había tratado con GDNF. Las
primeras parecen tener unas mayores proporciones de células dopaminérgicas si son tratadas adicionalmente con
GDNF. Las segundas, las NSCs, son mediadas por la secreción de factores
tróficos. Hay una tercera alternativa, las células
madre embrionarias (ESCs), que inducen neuronas con una gran proporción de
neuronas dopaminérgicas (4).
IV. Terapia génica
Puede así clasificarse en dos grupos: in vivo y ex vivo. En las primeras se usan vectores para la transmisión de
genes, tales como adenovirus, herpes simple, retrovirus y otros vectores no
virales, como liposomas(5).
Las estrategias de la terapia génica para EP son las siguientes: aumentar la concentración local de dopamina; ejercer efectos neuroprotectores
y reconstituyentes; mejorar el ambiente del sistema dopaminérgico de la
enfermedad y normalizar genéticamente a las células anormales.
3. Base molecular de la terapia génica
Se calcula que alrededor
del 80% de los pacientes con la EP tienen una causa idiopática, mientras que en
el 20% restante de los casos la causa es genética (6). En esta parte de la
revisión se incluye una síntesis que expone los diferentes factores
genéticos que influyen en la patogenia de la EP y de qué manera trascienden en
ella.
La EP es un trastorno
neurodegenerativo que forma parte de las denominadas sinucleinopatías (7).
Estas son la segunda causa más frecuente de la EP y se caracterizan porque la
alfa- sinucleína tiene tendencia a formar agregados anómalos en el citoplasma
de las neuronas y células gliales. Esta alfa-sinucleína es un componente
fundamental de los cuerpos de Lewy, al cual se atribuye un papel significativo
en la propagación de EP (8). Se cree que las conformaciones tóxicas de esta
proteína son oligómeros y protofibrillas, y se transmiten de unas células a
otras en forma de priones (esto explica la progresión de la enfermedad desde
zonas basales del cerebro hasta las áreas neocorticales). Además de esta
causa, en relación con esta proteína también encontramos su agregación con
beta-amiloide y tau.
Así pues, en esta
particular naturaleza de la alfa-sinucleína a formar agregados se han
encontrado mutaciones genéticas. El gen que codifica la alfa-sinucleína es el
SNCA, y las mutaciones aquí encontradas hasta el momento son el p.A30P, pE46K,
pH50Q, pG50D, pG51D, p.A53T, junto con duplicaciones y triplicaciones del gen.
Estas mutaciones son raras y entre ellas la más frecuente es el p.A53T (9).
Además de las mutaciones
encontradas en el SNCA se han encontrado mutaciones en el gen LRRK2,
responsable de EP de herencia dominante, de penetrancia incompleta y con
patología pleomórfica (10). Se han encontrado seis posibles mutaciones que
segregan enfermedad en el gen que da lugar a la proteína LRRK2 (cinco tipos
distintos de mutación con cambio de sentido y una supuesta mutación en sitio
de empalme) (11). La LRRK2 interviene en múltiples procesos celulares como la
autofagia, dinámica del citoesqueleto, cascada de quinasa, función
mitocondrial y tráfico vesicular. Las mutaciones en LRRK2 son la causa más
común de EP familiar de inicio tardío, y aunque aún no queda claro su papel
en la fisiología celular, este reciente hallazgo nos proporciona una mejor
perspectiva en la comprensión de la patogenia de EP.
Recientemente, también se
identificaron mutaciones en el gen VPS35, de incidencia muy baja y responsables
de EP de herencia dominante y de inicio tardío. Las mutaciones a este nivel
alteran el tráfico de catepsina D, proteína implicada en la degradación de
alfa-sinucleína. El VPS35, a través de endosomas y tráfico vesicular, tiene
vínculos con los genes SNCA y LRRK2.. En cuanto a las mutaciones que producen
EP de herencia recesiva y de inicio temprano están las que afectan a los genes
PARK2, PINK1, DJ-1, ATP13A2, PLA2G6 y FBX07.
Las mutaciones en PARKIN
2 son la causa más frecuente de la EP de herencia recesiva y de inicio
temprano debido a una microdelección de D6S305 (12). Entre los tipos de
mutaciones que tienen lugar a este nivel está el reordenamiento de exones y,
en amplia mayoría, las duplicaciones y delecciones de exones. La patología
que produce el PARKIN 2 es la de pérdida de neuronas en la sustancia negra.
La segunda causa
genética más frecuente de EP de herencia recesiva es la que tiene lugar en el
gen PINK1 (13). En relación con la forma familiar del Parkinson, encontramos
el cromosoma 1p36 (PARK6) y las mutaciones en PINK1 están asociadas con este
último. Concretamente, PINK1 se encuentra en la mitocondria de las células
ejerciendo una función protectora sobre esta. Cuando dicha función se altera,
se produce estrés celular, el cual parece estar relacionado con el desarrollo
del Parkinson hereditario. Esta tiene la capacidad de fosforilar a PARKIN2 con
el fin de regular la mitofagia a nivel mitocondrial.
Las mutaciones en DJ-1
son extremadamente raras, y la proteína que codifica –DJ- tiene un papel
relevante en la protección de las células dopaminérgicas frente al estrés
oxidativo, así como en el mantenimiento de la circulación normal de dopamina
en la vía nigroestriatal (14). Además, parece tener una actividad semejante a
la de las proteínas chaperonas, puede inhibir la agregación de
alfa-sinucleínas y llevar a cabo transcripciones de genes neuroprotectores y
antiapoptóticos. De esta manera, protege a las células neuronales de la
apoptosis en cuanto están en un ambinte de estrés oxidativo.
Otras formas hereditarias
recesivas de la EP, pero más raras, son las que están causadas por mutaciones
en tres genes: ATP13A2, PLA2G6, y FBXO7.
Las mutaciones en el gen
de la glucocerebrosidasa (GBA1) causan la enfermedad de Gaucher, cuyos
portadores no solo tienen un mayor riesgo a desarrollar EP, sino que también
presentan síntomas muy parecidos a los de la EP idiopática. Se ha observado
que la mutación en el gen GBA1 conlleva una disminución de la actividad de la
b- glucocerebrosiledasa, lo que provoca una disfunción lisosomal y
acumulación del sustrato glucosilceramida. Este hecho llevó a revisar el
vínculo que había entre ambas enfermedades y los factores que hacen que la
alfa-sinucleína monomérica se transformé hacia formas neurotóxicas y
agregados oligoméricas que contribuyen a la progresión de la enfermedad (15).
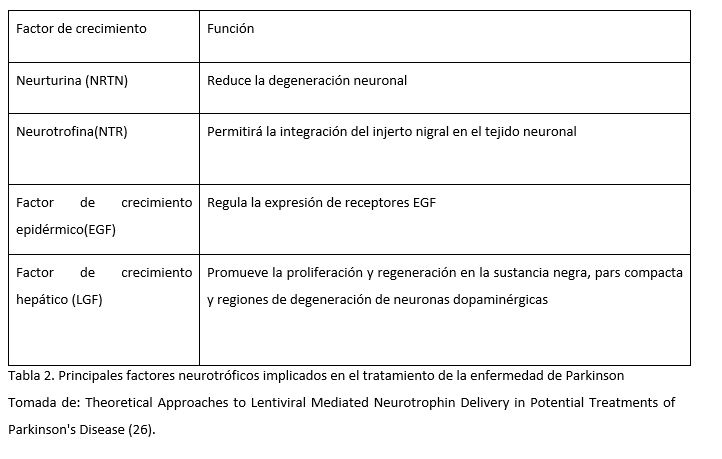
Por último, cabe
mencionar que los factores neurotróficos derivados de la línea celular glial
(GDNF) y la neurturina son muy importantes en el mantenimiento de la
supervivencia de las células neuronales, y que en los pacientes con la EP
estos se encontraban desregulados y disminuidos al igual que sus receptores
(9).
4. Terapia génica
La terapia génica es una técnica terapéutica en la que se inserta un gen funcional en las células de un paciente para corregir un defecto genético que suele causar patología, se puede realizar por medio de virus que transportan esas moléculas terapéuticas (16). Actualmente, existen dos tipos de métodos de terapia génica: la terapia génica sintomática y la terapia génica centrada en la modificación de la enfermedad (17). En el caso de la terapia génica sintomática, hablamos principalmente del transporte del enzima dopa-decarboxilasa, la AADC(que transforma la levodopa en dopamina) aumentando en el cerebro, por tanto, la dopamina, que es deficitaria en los enfermos con Parkinson (18). Esto se ha probado en animales en los que dosis más bajas de levodopa significaban dosis más altas con transporte génico de AADC, dando un efecto antiparkinsoniano. También se han usado lentivirus para transportar tirosina hidroxilasa, AADC y GTP ciclohidrolasa, cuya combinación da lugar a la dopamina. La unión de lentivirus y dopamina ha sido probado en animales dando, de igual manera, un beneficio antiparkinsoniano.
Por otra parte, el segundo método, se centra en la familia de ligandos de células gliales, los llamados GDNF y neurturina. El transporte génico de estos factores tróficos, como la neurturina, protege el circuito nigroestriatal y la función motora (pero solo fue probado en roedores y primates no humanos). Un aspecto de relevancia fue el hecho que dicho transporte génico presentó una gran eficiencia en los ensayos preclínicos, pero fue muy ineficiente al extrapolarlo a enfermos de EP, se cree que esto fue debido a la extensa degeneración del sistema nigroestriatal al tiempo que los pacientes son tratados y además, las fibra nerviosas que quedan son defectuosas en los mecanismos de transporte axonal, por lo que los factores tróficos no son llevados eficientemente (19).
La terapia génica en la EP es todavía una gran incógnita,
pues ningún estudio ha demostrado aún un beneficio clínico sólido
en humanos. Aunque es posible una solución, actualmente, solo se ha usado la
terapia génica en esta enfermedad para síntomas motores,
pero para otros síntomas como los psiquiátricos, se necesitará una terapia génica diferente.
5. Nuevas alternativas terapeúticas
En lo que al tratamiento de la EP respecta,
los últimos avances proponen una imitación de la naturaleza para así poder
avanzar en este sentido: la administración de factores neurotróficos mediante
retrovirus, es una alternativa muy positiva para conseguir administrar estos
factores a nuestras células diana.
En la presente revisión
pretendemos abordar las principales terapias en el tratamiento de la EP. Cabe
resaltar que, a diferencia de los tratamientos introducidos hasta el momento,
esta nueva perspectiva pretende actuar sobre la patogenia de la enfermedad para
así buscar un tratamiento efectivo y no meramente sintomático.
Los factores neurotróficos son compuestos biológicos que estimulan la
regeneración celular y, por lo tanto, facilitan el proceso de curación. Para
ejercer su acción se unen a receptores celulares y, posteriormente, activan las
cascadas de señalización celular que regulan la mitosis, la diferenciación y la
apoptosis (20).
NRTN es un análogo natural de los factores neurotróficos derivados de la
línea celular glial (GDNF), comúnmente utilizado para atacar a las neuronas
dopaminérgicas en animales con EP (21). Sus efectos observados incluyen el
retraso de la degeneración de las neuronas, la protección selectiva de las
neuronas dopaminérgicas y la mejora general del funcionamiento neuronal. La
administración de este factor fue probado por Gasmi et al en 2007, se
realizaron ensayos con animales para lograr el suministro estriatal de NRTN
utilizando CERE-120 (virus adenoasociado tipo 2 – AAV2). Desde entonces, la
administración de este factor, ha supuesto una alternativa muy prometedora en
el tratamiento de la EP.
Con respecto a NTR, se ha demostrado que tiene efectos beneficiosos en la EP
idiopática, gracias a su efecto integrador de injertos en la sustancia
negra (22).
Los estudios de Iwakura, desvelaron que existía un déficit en la expresión de EGF en los cerebros
post mortem de los pacientes con EP. De esta forma, se pensó que la
administración de este factor, sería una alternativa prometedora en el
tratamiento de la EP (23).
Gobernado et al. administró LGF en ratas y observó una posible actividad
neuroprotectora. LGF es un mitógeno hepático que promueve la proliferación de
varios tipos de células y facilita la regeneración de tejidos. Tras la
aplicación periférica de LGF en cuerpo estriado izquierdo, aumentó de forma
unilateral la brotación de los terminales positivos para la tirosina
hidroxilasa y la expresión del transportador de dopamina. LGF también estimuló
la fosforilación y regulación de proteínas críticas para la supervivencia
celular, incluidos Bcl2 y Akt (24).
Haciendo
referencia a la administración de genes basados en retrovirus, podemos decir
que el objetivo de estos sistemas de administración
es hacer llegar genes a las células diana, utilizando como medio de transporte
virus, en este caso utilizaremos lentivirus (25). Una vez que los genes
objetivo se empaquetan en vectores virales, convierten su ARN monocatenario en
un ADN bicatenario que puede integrarse de manera estable en el genoma del
huésped. El vector integrado, llamado provirus, sufre replicación y
transcripción en el genoma del hospedador, lo que produce los ARNm virales y
también el ARN empaquetado.
Cabe destacar la
importancia de controlar el uso no tan noble de estos sistemas, como la
sobreexpresión de genes.
6. Conclusión
Actualmente, podemos clasificar a la enfermedad del Parkinson como una patología con una etiología multifactorial, así como con una gran influencia genética. En nuestros días, las terapias existentes se centran casi exclusivamente en la sintomatología, sin embargo, los estudios que se están llevando a cabo con terapia génica suponen una alternativa dirigida directamente a la raíz del problema. A través de los argumentos que se han ido refiriendo a lo largo de la revisión, se puede llegar a la conclusión de que la terapia génica puede resultar beneficiosa en el tratamiento de esta enfermedad, aunque esto no haya sido concluyente en la totalidad de los artículos revisados. Numerosas son las vías que aún quedan por explorar en este campo de la genética, lo cual augura un futuro esperanzador para la enfermedad del Parkinson.
7. Conflicto de intereses
Los autores de la
revisión científica declaran no presentar ningún tipo de conflicto de
intereses.
8. Referencias
- Yashura T, Kameda M, Agari T, Date I. Regenerative Medicine for Parkinson’s Disease. Neurol Med Chir. 2015; 55: 113-123.
- Lindvall O, Rehncrona S, Gustavii B, et al. Fetal dopamine-rich esencephalic grafts in Parkinson’s disease. Lancet. 1998; 2: 1483–1484.
- Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med. 2005; 11: 703–704.
- Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000; 28: 31–40.
- Yasuhara T, Date I. Gene therapy for Parkinson’s disease. J Neural Transm Suppl. 2009; 73: 01–309.
- Bhat S, Acharya UR, Hagiwara Y, Dadmehr N, Adeli H. Parkinson’s disease: Cause factors, measurable indicators, and early diagnosis. Comput Biol Med. 2018; 102: 234–41.
- Toulorge D, Schapira AHV, Hajj R. Molecular changes in the postmortem parkinsonian brain. J Neurochem. 2016; 139: 27–58.
- Schulz JB, Hausmann L, Hardy J. 199 years of Parkinson disease – what have we learned and what is the path to the future? J Neurochem. 2016; 139: 3–7.
- Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: Mendelian versus non‐Mendelian inheritance. J Neurochem. 2016; 139: 59–74.
- Weisenhorn DMV, Giesert F, Wurst W. Diversity matters – heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s Disease. J Neurochem. 2016; 139: 8–26.
- Goedert M, Jakes R, Anthony Crowther R, Grazia Spillantini M. Parkinson’s Disease, Dementia with Lewy Bodies, and Multiple System Atrophy as alpha-Synucleinopathies. Methods Mol Med. 2001; 62: 33–59.
- Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004; 44: 601–607.
- Matsumine H, Yamamura Y, Hattori N, et al. A microdeletion of D6S305 in a family of autosomal recessive juvenile parkinsonism (PARK2) Genomics. 1998; 49: 143–146.
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004; 304: 1158–1160.
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003; 299: 256–259.
- Man JHK, Groenink L, Caiazzo M. Cell reprogramming approaches in gene- and cell-based therapies for Parkinson’s disease. J Control Release. 2018; 286: 114–24.
- Kordower JH, Bjorklund A. Trophic factor gene therapy for Parkinson’s disease. Mov Disord. 2013; 28(1): 96-109.
- Mittermeyer G, Christine CW, Rosenbluth KH, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther. 2012; 23(4): 377-381.
- Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136(Pt 8). 2013; 136(Pt 8): 2419–2431.
- Barker RA. Parkinson’s disease and growth factors — are they the answer?. Parkinsonism Relat Disord. 2009; 15(Suppl 3): 181–184.
- Ye M, Wang XJ, Zhang YH, et al. Transplantation of bone marrow stromal cells containing the neurturin gene in rat model of Parkinson’s disease. Brain Res. 2007; 1142: 206–216.
- Mogi M, Togari A, Kondo T, et al. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1999; 270(1): 45–48.
- Iwakura Y, Piao YS, Mizuno M, et al. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: Neurotrophic implication in nigrostriatal neurons. J Neurochem. 2005; 93(4): 974–983.
- Gonzalo-Gobernado R, Calatrava-Ferreras L, Reimers D, et al. Neuroprotective Activity of Peripherally Administered Liver Growth Factor in a Rat Model of Parkinson’s Disease. PLoS One. 2013; 8(7): 67771.
- Palfi S, Gurruchaga JM, Lepetit H, et al. Long-Term Follow-Up of a Phase I/II Study of ProSavin, a Lentiviral Vector Gene Therapy for Parkinson’s Disease. Hum Gene Ther Clin Dev. 2018; 29(3): 148–55.
- Qudrat A, Unni N. Theoretical Approaches to Lentiviral Mediated Neurotrophin Delivery in Potential Treatments of Parkinson’s Disease. Yale J Biol Med. 2016; 89(2): 215–25.
Therapeutic Action and Gene Therapy for Parkinson’s Disease
There are numerous therapeutic alternatives for the treatment of Parkinson’s disease (PD). However, none of this can stop the progression of this disease, although they do have major symptomatic benefit. Thus, interest arises in new therapeutic alternatives such as cell transplantation, glial cell line-derived neurotrophic factor (GDNF) infusion, cell therapy or gene therapy. This review presents a general overview of current therapies used for PD and future lines of research, highlighting the role of gene therapy. The different gene therapy approaches for PD are revised. These include a symptomatic gene therapy, which aims to restore dopamine levels in the brain, and a therapy focused on the modification of the disease, for which the glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) plays a fundamental role. Finally, the authors conclude that these lines of research are still far from clinical application.
Keywords: glial cell line-derived neurotrophic factor, cell therapy, gene therapy, glial cells.
- Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease. It is a pathology in which the nigrostriatal pathway is affected, with a characteristic deficit of dopamine production. PD is characterized by motor symptoms such as freezing, postural instability, rigidity, or tremor, as well as non-motor symptoms, including anxiety, dementia and depression. In this review, the existing therapies for PD are described, with special emphasis on gene therapy, which has promising results to date. In gene therapy, viral vectors are employed to deliver genetic material that will modify the cause of the disease.
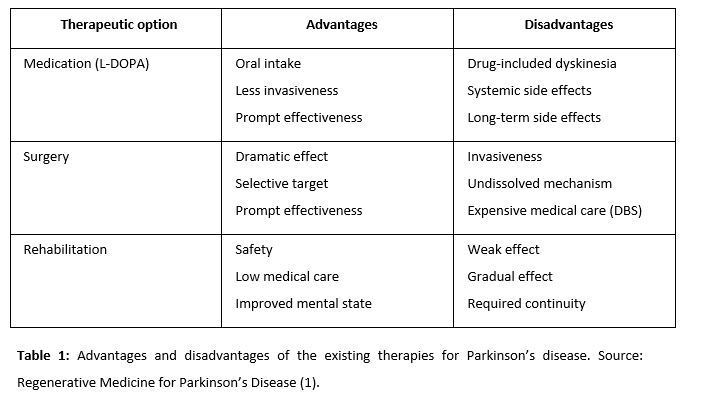
- Therapeutic action for Parkinson’s disease
The basis of the pathogenesis of PD is progressive degeneration of dopaminergic neurons in the nigrostriatal system. This makes PD a good target for regenerative medicine (1).
2.1. Historical Development of Regenerative Medicine for PD
2.1.1. Fetal nigral cell transplantation
Fetal nigral cells were transplanted to make new synaptic connections and to synthesize dopamine (2). At the outset of the 21st century, studies revealed insufficient functional recovery and increased dopamine uptake in elderly patients and some patients with tardive dyskinesia. Other studies reported more optimistic aspects of fetal nigral cell transplantation using a cell suspension rather than solid tissue. Patients showed improvement in motor function and a reduced immune response in dyskinesia. Thus, fetal nigral cell transplantation might exert strong therapeutic effects for a long time with appropriate patient selection.
2.1.2. Infusion of glial cell line-derived neurotrophic factor (GDNF)
Alternatively, the intraparenchymal injection of GDNF can have important therapeutic effects, especially if the injection is at the intrastriatal level (3). was demonstrated as another hopeful therapeutic option for PD patients. Moreover, unilateral GDNF administration has been proved to improve bilateral function.
2.1.3. Cell therapy
One cell-based strategy is to transplant encapsulated cells, which allows combining several genetically modified cell types. Cells inside the capsule survived for up to 6 months in vivo with enough oxygen, nutrients, and a semi-permeable membrane. The capsule protects cells from immunological rejection and tumor formation. Neural stem cells (NSCs) and mesenchymal stem cells (MSCs) proved to be good candidates for this therapy, as the number of NSCs that survived in vivo was higher if they had previously been treated with GDNF. GDNF treatment increased the proportion of dopaminergic neurons in MSCs. NSCs were mediated by secreted trophic factor. Embryonic stem cells (ESCs), the last alternative, induce neurons with a high proportion of dopaminergic neurons (4).
2.1.4. Gene therapy
Gene therapy is classified into two groups: in vivo and ex vivo. Direct gene delivery using vectors is the main characteristic of in vivo gene therapy. Herpes simplex virus (HSV), retrovirus, adenovirus, and other non-viral vectors such as liposome are used (5).
The purposes of gene therapy are to increase local dopamine concentration, to exert neuroprotective/neurorestorative effects, to ameliorate the microenvironment of the dopaminergic system involved in PD, and to normalize genetically abnormal cells.
- Biological basis of gene therapy
Around 80% of patients with PD are considered as idiopathic, whereas the remaining 20% cases are presumed to be genetic. In this section, the different genetic factors that influence the pathogenesis of PD are presented.
PD is a neurodegenerative disorder and the most common synucleinopathy (7). It is characterized by the accumulation of anomalous alpha-synuclein aggregates in the cytoplasm of neurons and glial cells. Alpha-synuclein is a fundamental component of Lewy bodies, which play a significant role in the progression of PD. It is believed that alpha-synuclein oligomers and fibrils are toxic. The protein has “prion-like” properties that can spread throughout a network of neurons. This explains the progression of the disease from the basal ganglia to the neocortex. Moreover, alpha-synuclein aggregates with beta-amyloid and tau. However, mutations responsible for the disease were identified in the alpha-synuclein gene. Highly penetrant mutations producing rare, monogenic forms of the disease have been discovered in singular genes such as SNCA. The first identified missense mutation, p.A53T is the most frequent (9), the rest are p.A30P, pE46K, p.H50Q, p.G50D and p.G51D. In addition to SNCA, autosomal-dominant PD-causing mutations with pleomorphic pathology and incomplete penetrance have been found in the gene encoding Leucine-rich repeat kinase 2 (LRRK2) (10). Six mutations in LRRK2 have been identified (five types of missense mutation and one splicing mutation) (11). This protein is involved in numerous cellular processes including autophagy, cytoskeletal dynamics, kinase cascades, mitochondrial function and vesicular trafficking. Overall, mutations in LRRK2 are the most commonly known genetic cause of late-onset familial PD. Although the role of LRRK2 in cellular physiology is still unclear, this recent finding allows for a better understanding of the pathogenesis of PD.
Recently, mutations were also identified in the VPS35 gene, responsible for autosomal dominant late-onset PD. The mutant VPS35 protein alters the trafficking of cathepsin D, a protein involved in the degradation of alpha-synuclein. VPS35 is linked to the other autosomal dominant PD genes SNCA and LRRK2 through endosomes and vesicular trafficking. Mutations in genes PARK2, PINK1, DJ-1, ATP13A2, PLA2G6 and FBX07 have been shown to cause autosomal recessive PD and early-onset. Mutations in PARK2 are the most frequent I of autosomal recessive and early-onset PD due to a microdeletion of D6S305 (12). These mutations consist of exon rearrangements, including both deletions and duplications. The pathology caused by PARK2 consists of severe neuronal loss in the substantia nigra.
Mutations in PINK1 are the second most common cause of autosomal recessive PD (13). The PARK6 locus was first mapped on chromosome 1p36 and mutations in PINK1 are associated with this chromosome. PINK1 is located in the mitochondria of cells and it helps protect mitochondria from malfunctioning during periods of cell stress. When this function is altered, cellular stress occurs, which seems to be related to the development of familial PD. PINK1 phosphorylates PARKIN2 to regulate mitophagy of damaged mitochondria.
Mutations producing rare forms of the disease have been discovered in gene DJ-1. This gene has been reported to protect dopaminergic cells against oxidative stress and to play a key role in maintaining normal dopaminergic function in the nigrostriatal pathway (14). Furthermore, DJ-1 seems to have chaperone activity and the ability to inhibit alpha-synuclein aggregation. It may be involved in transcriptional regulation of neuroprotective and anti-apoptotic genes and in the protection of neurons from oxidative stress induced apoptosis.
Other rare, recessively inherited forms of PD are caused by mutations in three genes: ATP13A2, PLA2G6, y FBXO7. Mutations in GBA1, the gene encoding for the lysosomal enzyme glucocerebrosidase, cause Gaucher disease. Patients suffering from this disease have higher risk of developing PD and have symptoms associated with idiopathic PD. It was observed that mutation in gene GBA1 lead to a loss of beta-glucocerebrosidase activity, which causes lysosomal dysfunction and an increased substrate glucosylceramide accumulation. This led to review the link between the two diseases and the factors that cause monomeric alpha-synuclein to transform into neurotoxic forms and oligomeric aggregates that contribute to the progression of the disease (15).
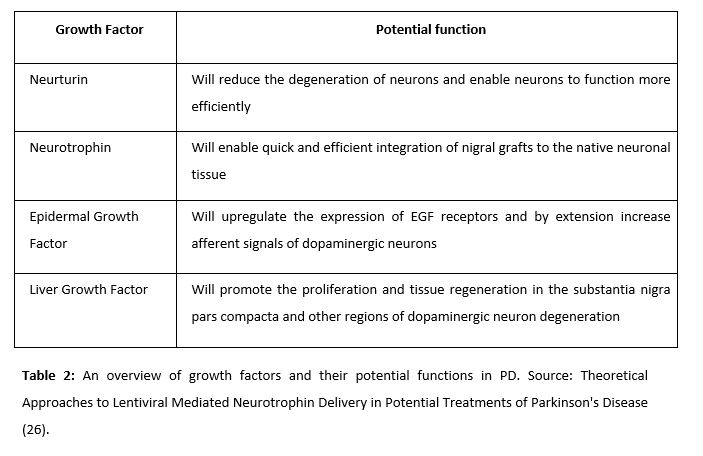
Finally, it is worth noting the importance of the glial cell line-derived neurotrophic factor (GDNF) and neurturin for the survival of neuronal cells. These, as well as their receptors, were decreased and deregulated in patients with PD (9).
- Gene therapy
Gene therapy is a therapeutic technique in which a functional gene is inserted into a patient’s cells. Its aim is to correct a genetic defect that often causes pathology. It can be performed by using viruses that carry these therapeutic molecules (16). Nowadays, there are two different gene therapy methods: symptomatic gene therapy and gene therapy focused on disease modification (17). Symptomatic gene therapy mainly refers to the transport of the dopa-decarboxylase enzyme, AADC (which transforms levodopa into dopamine). This increases dopamine in the brain, which is deficient in patients with PD (18). This has been tested in animals where lower doses of levodopa meant higher doses with gene transport of AADC, producing an antiparkinsonian effect. Lentiviruses have also been used to transport tyrosine hydroxylase, AADC, and GTP cyclohydrolase, the combination of which results in dopamine. The combination of lentiviruses and dopamine has been tested in animals also having an antiparkinsonian effect.
The second method focuses on GDNF and neurturin. The gene transport of these trophic factors, such as neurturin, protects the nigrostriatal pathway and the motor function. Nonetheless, this was not tested in humans but only in rodents and primates. Quite significantly, the gene transport was highly efficient in preclinical tests, but it was very inefficient when applied to PD patients. This could be due to the extensive degeneration of the nigrostriatal system during treatment. Furthermore, the remaining nerve fibers are defective in the axonal transport mechanisms, so trophic factors are not carried efficiently (19).
Gene therapy in PD is still a mystery, as no study has yet shown undisputed clinical benefit in humans. Although a solution is possible, currently gene therapy has only been used to treat motor symptoms. However, other symptoms such as psychiatric ones would require a different type of gene therapy.
- New therapeutic alternatives
Regarding the treatment of PD, the latest advances propose an imitation of nature. The distribution of neurotrophic factors using retroviruses is a very positive alternative to provide our target cells with these factors.
In this review we intend to address the main therapies for the treatment of PD. It should be noted that, unlike the treatments introduced so far, this new perspective aims to act on the pathogenesis of the disease in order to find an effective treatment instead of a merely symptomatic one.
Neurotrophic factors are biological compounds that stimulate cell regeneration and, therefore, ease the healing process. They function by binding to cellular receptors and subsequently activating the cell signaling cascades that regulate mitosis, differentiation, and apoptosis (20).
Neurturin (NRTN) is a natural analogue of neurotrophic factors derived from the glial cell line (GDNFs), commonly used to attack dopaminergic neurons in PD animals (21). Its observed effects include delayed degeneration of neurons, selective protection of dopaminergic neurons, and general improvement of neural functioning. The administration of this factor was tested by Gasmi et al in 2007. Animal trials were conducted to achieve striatal delivery of NRTN by using CeRe-120 (an adeno-associated virus type 2 – AAv2). Since then, the administration of this factor has been a very promising alternative for the treatment of PD.
NRTN has been proved to have beneficial effects in idiopathic PD, thanks to its integrating effect of grafts in the substantia nigra (22).
Iwakura’s studies revealed that there was a deficit in the expression of EGF in post-mortem brains of patients with PD. Thus, it was thought that the administration of this factor would be a promising alternative in the treatment of the disease (23). Gobernado et al. administered LGF in rats and observer a possible neuroprotective activity. LGF is a hepatic mitogen that promotes proliferation of various cell types and facilitates tissue regeneration. Upon peripheral application of LGF to the left striatum, sprouting of tyrosine hydroxylase-positive terminals and dopamine transporter expression was unilaterally increased. LGF also stimulated the phosphorylation and regulation of proteins critical for cell survival – including Bcl2 and Akt.
The aim of the administration of genes based on retroviruses is to deliver genes to the target cells, using viruses as means of transport. In this case, lentiviruses are used (25). Once the target genes are packaged into these viral vectors, they convert their single stranded RNA into a double stranded DNA that can stably integrate into the host genome. The integrated vector, called provirus, undergoes replication and transcription in the host genome, producing both viral mRNAs and packaged RNA. The integrated vector, called the provirus, undergoes replication and transcription in the host genome producing the viral mRNAs and the packaged RNA as well.
The importance of controlling the not-so-noble use of these systems, such as overexpression of genes, should be emphasized.
- Conclusion
Today, we can classify PD as a pathology with a multifactorial etiology and a strong genetic influence. Nowadays, the existing therapies focus almost exclusively on the symptomatology. However, recent studies on gene therapy seek to provide an alternative that goes directly to the root of the problem. Through the arguments exposed in this review, it can be concluded that gene therapy may be beneficial for the treatment of PD, although not all the articles reviewed conclude this. There are still numerous aspects to be explored in the field of genetics, which appears to indicate a promising future for the treatment of PD.
Conflicts of Interest Statement
The authors declare that there are no conflicts of interest in this article.
References
- Yashura T, Kameda M, Agari T, Date I. Regenerative Medicine for Parkinson’s Disease. Neurol Med Chir. 2015; 55: 113-123.
- Lindvall O, Rehncrona S, Gustavii B, et al. Fetal dopamine-rich esencephalic grafts in Parkinson’s disease. Lancet. 1998; 2: 1483–1484.
- Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med. 2005; 11: 703–704.
- Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000; 28: 31–40.
- Yasuhara T, Date I. Gene therapy for Parkinson’s disease. J Neural Transm Suppl. 2009; 73: 01–309.
- Bhat S, Acharya UR, Hagiwara Y, Dadmehr N, Adeli H. Parkinson’s disease: Cause factors, measurable indicators, and early diagnosis. Comput Biol Med. 2018; 102: 234–41.
- Toulorge D, Schapira AHV, Hajj R. Molecular changes in the postmortem parkinsonian brain. J Neurochem. 2016; 139: 27–58.
- Schulz JB, Hausmann L, Hardy J. 199 years of Parkinson disease – what have we learned and what is the path to the future? J Neurochem. 2016; 139: 3–7.
- Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: Mendelian versus non‐Mendelian inheritance. J Neurochem. 2016; 139: 59–74.
- Weisenhorn DMV, Giesert F, Wurst W. Diversity matters – heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s Disease. J Neurochem. 2016; 139: 8–26.
- Goedert M, Jakes R, Anthony Crowther R, Grazia Spillantini M. Parkinson’s Disease, Dementia with Lewy Bodies, and Multiple System Atrophy as alpha-Synucleinopathies. Methods Mol Med. 2001; 62: 33–59.
- Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004; 44: 601–607.
- Matsumine H, Yamamura Y, Hattori N, et al. A microdeletion of D6S305 in a family of autosomal recessive juvenile parkinsonism (PARK2) Genomics. 1998; 49: 143–146.
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004; 304: 1158–1160.
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003; 299: 256–259.
- Man JHK, Groenink L, Caiazzo M. Cell reprogramming approaches in gene- and cell-based therapies for Parkinson’s disease. J Control Release. 2018; 286: 114–24.
- Kordower JH, Bjorklund A. Trophic factor gene therapy for Parkinson’s disease. Mov Disord. 2013; 28(1): 96-109.
- Mittermeyer G, Christine CW, Rosenbluth KH, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther. 2012; 23(4): 377-381.
- Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136(Pt 8). 2013; 136(Pt 8): 2419–2431.
- Barker RA. Parkinson’s disease and growth factors — are they the answer?. Parkinsonism Relat Disord. 2009; 15(Suppl 3): 181–184.
- Ye M, Wang XJ, Zhang YH, et al. Transplantation of bone marrow stromal cells containing the neurturin gene in rat model of Parkinson’s disease. Brain Res. 2007; 1142: 206–216.
- Mogi M, Togari A, Kondo T, et al. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1999; 270(1): 45–48.
- Iwakura Y, Piao YS, Mizuno M, et al. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: Neurotrophic implication in nigrostriatal neurons. J Neurochem. 2005; 93(4): 974–983.
- Gonzalo-Gobernado R, Calatrava-Ferreras L, Reimers D, et al. Neuroprotective Activity of Peripherally Administered Liver Growth Factor in a Rat Model of Parkinson’s Disease. PLoS One. 2013; 8(7): 67771.
- Palfi S, Gurruchaga JM, Lepetit H, et al. Long-Term Follow-Up of a Phase I/II Study of ProSavin, a Lentiviral Vector Gene Therapy for Parkinson’s Disease. Hum Gene Ther Clin Dev. 2018; 29(3): 148–55.
- Qudrat A, Unni N. Theoretical Approaches to Lentiviral Mediated Neurotrophin Delivery in Potential Treatments of Parkinson’s Disease. Yale J Biol Med. 2016; 89(2): 215–25.
AMU 2019. Volumen 1, Número 1
Fecha de envío: Fecha de aceptación: Fecha de publicación:
15/04/2019 22/04/2019 31/05/2019
Cita el artículo: Rocobado-Pozo M.Y, Ruiz-Medina M, Reina-Reina I, Reyes-Molina A, Durán-Ávila J.J. Actuaciones terapéuticas y terapia génica en la enfermedad de Parkinson. AMU. 2019; 1(1):56-75